Type-I Interferons in Alzheimer's Disease and Other Tauopathies
 Sophie A. I. Sanford
Sophie A. I. Sanford William A. McEwan
William A. McEwan- 1Department of Clinical Neurosciences at the University of Cambridge, Cambridge, United Kingdom
- 2UK Dementia Research Institute at the University of Cambridge, Cambridge, United Kingdom
The detection of pathogen-associated molecular patterns can elicit the production of type-I interferons (IFNs), soluble cytokines that induce a transcriptional state inhibitory to viral replication. Signatures of type-I IFN-driven gene expression, and type-I IFNs themselves, are observed in the central nervous system during neurodegenerative diseases including Alzheimer's disease and other tauopathies, the umbrella term for diseases that feature aggregation of the cytosolic protein tau. The contribution of the type-I IFN response to pathological progression of these diseases, however, is not well-understood. The wholesale transcriptional changes that ensue from type-I IFN production can both promote protective effects and lead to damage dependent on the context and duration of the response. The type-I IFN system therefore represents a signaling pathway with a potential disease-modifying role in the progression of neurodegenerative disease. In this review we summarize the evidence for a type-I IFN signature in AD and other tauopathies and examine the role of aggregated proteins as inflammatory stimuli. We explore both the protective role of IFN against protein pathologies as well as their downstream toxic consequences, which include the exacerbation of protein pathology as a potentially destructive feed-forward loop. Given the involvement of type-I IFNs in other neurogenerative diseases, we draw comparisons with other categories of homotypic protein aggregation. Understanding how type-I IFN influences progression of AD and other tauopathies may yield important insight to neurodegeneration and identify new targets in an area currently lacking disease-modifying therapies.
Introduction
Alzheimer's disease (AD) is the most common form of dementia and is anticipated to affect more than 113 million people worldwide by 2050 (Knopman et al., 2021). AD is characterized by two distinct pathologies in the post-mortem brain (Stelzmann et al., 1995). Plaques of beta-amyloid (Aβ) peptide, a cleavage product of the transmembrane protein, amyloid precursor protein (APP), accumulate in the extracellular spaces of the brain. In addition, fibrillar and hyperphosphorylated assemblies of the microtubule-associated protein tau accumulate in the cytoplasm of neurons (Goedert and Spillantini, 2006). Mutations in APP can lead to dominantly inherited, early-onset variants of AD, though these inherited forms make up <1% of AD cases (Laurent et al., 2018). The “amyloid cascade hypothesis” places Aβ pathology as an upstream, causative insult that unleashes a range of ensuing consequences including tau pathology and neurotoxicity (Hardy and Higgins, 1992). However, clinically targeting Aβ has so far failed to yield cognitive benefit (Karran and De Strooper, 2022). This has directed focus toward other targets such as tau lesions, which correlate strongly with cognitive decline (Nelson et al., 2007).
Tau assemblies are present in a range of neurodegenerative diseases alongside AD, classed as tauopathies. Several non-synonymous point mutations in MAPT, the gene that encodes tau, give rise to familial inherited tauopathies such as frontotemporal lobar degeneration with tau-immunoreactive inclusions (FTLD-tau) (Goedert, 2018). These findings establish tau as a causative factor in pathological progression, at least in these rare diseases and potentially more broadly in the tauopathies. In Pick's disease (PiD), progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD), tau pathology is the main, often sole observed protein pathology (Spillantini and Goedert, 2013). The mechanism that leads to the assembly of tau during neurodegeneration is uncertain. Cell-autonomous mechanisms likely drive the earliest tau pathology, which is apparent in the majority of human brains by the age of 30 years (Braak and Del Tredici, 2015). Misfolded tau may also act in a “prion-like” manner promoting its propagation through iterative rounds of seeded aggregation. Injection of mouse brains with tau assemblies can induce tau aggregation in the neurons of the recipient animal (Clavaguera et al., 2009; De Calignon et al., 2012; Liu et al., 2012a). Similar results can also be obtained in cell-based and ex vivo models (Frost et al., 2009; Guo and Lee, 2011; McEwan et al., 2017; Miller et al., 2021). While the contribution of this process to disease progression remains undetermined, a unifying feature of AD and several tauopathies is inflammation.
Microglia are a critical component of the brain's innate immune response, the first line of defense against foreign pathogens during infection. This response limits the early replication of pathogens while adaptive immune responses are generated (Le Bon and Tough, 2002). Pathogen-associated molecular patterns (PAMPs) or host-derived danger-associated molecular patterns (DAMPs) are recognized by germ-line encoded pattern recognition receptors (PRRs) on the surface of, and inside, host cells (Iwasaki and Medzhitov, 2004; Roh and Sohn, 2018). Microglia are the major site of PRR expression in the brain, though other cell types, particularly astrocytes, also contribute. Engagement of PRRs can result in the transcription of cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6), tumor necrosis factor (TNF) and type-I interferons. Each of these have specific roles, including recruitment of professional immune cells and the orchestration of the adaptive immune response. Type-I IFNs comprise IFN-α, of which there are 12 to 13 subtypes, and IFN-β and as well as IFN-ε, IFN-κ, IFN-ω, IFN-δ, IFN-ζ, and IFN-τ. The type-I IFNs have a critical role in inducing an antiviral state in infected and neighboring cells. They exert this function by binding to the type I IFN receptor complex (IFNAR) and initiating signaling through kinases JAK1 and TYK2 which in turn phosphorylate STAT1 and STAT2 transcription factors (Figure 1). This leads to the upregulation of around 2,000 genes in humans, a response that is conserved across mammals (Shaw et al., 2017). As well as their antiviral effects, type-I IFNs have a further role in dampening pro-inflammatory cytokines (Prinz et al., 2008; Goldmann et al., 2016). Each of the major cell types of the CNS retain the ability to both produce and respond to type-I IFN (Supplementary Tables 1–3), though astrocytes and microglia are considered the major sources of production (Scheu et al., 2019).
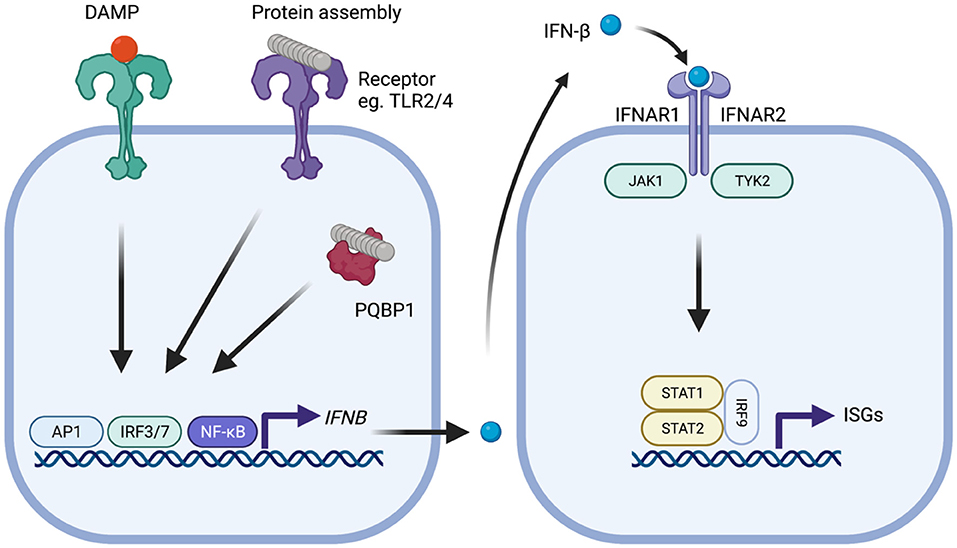
Figure 1. Type-I IFN signaling in the context of neurodegenerative disease. Protein assemblies such as β-amyloid and tau activate innate immune signaling through interactions with cell surface receptors such as TLR2 and TLR4 or intracellular receptors such as PQBP1. Damage associated molecular patterns similarly provoke activation of signaling. Activation of downstream signaling pathways, notably activator protein-1 (AP1), interferon regulatory factor 3 (IRF3), or IRF7 and nuclear factor-kappa B (NF-kB), leads to production of IFN-β which subsequently binds to the type-I IFN receptor complex comprising IFNAR1 and IFNAR2 that is expressed on all nucleated cells. Following type-I IFN binding, the IFNAR receptor complex initiates signaling through the adaptor kinases JAK1 and TYK2 leading to activation of STAT1/STAT2/IRF9 heteromultimers. This complex, referred to as ISGF3, migrates to the nucleus and induces transcription of interferon-stimulated genes (ISGs) that possess interferon-sensitive response element (ISRE). The protein products of these genes help establish an antiviral state and include several innate immune sensors.
Inflammation is a key feature of the degenerating brain. In AD, inflammatory markers, including TNF, IL-1β and type-I IFN precede the appearance of symptoms (Taylor et al., 2014; Taipa et al., 2019) and chronically activated, dysfunctional microglia are widespread in the post-mortem brain (Tarkowski, 2003; Navarro et al., 2018). Reactive microglia can be found associated with sites of both tau and Aβ pathologies (Serrano-Pozo et al., 2011). In tauopathies that do not feature Aβ plaques, activated microglia increase proportionately with tau pathology and are found in close proximity to sites of aggregation (Paulus et al., 1993; Gerhard et al., 2006; Malpetti et al., 2020). It is now understood that prolonged immune activation can exacerbate protein aggregation in AD and tauopathies [reviewed by Wyss-Coray and Mucke (2002) and Laurent et al. (2018)]. As major players in the response to infection, and in the degenerating brain, the role of type-I IFNs has come under scrutiny.
Type-I IFN Signature of Alzheimer's Disease and Tauopathies
Transcripts of IFNα/β and downstream IFN-stimulated genes (ISGs), are significantly higher in brains of AD and clinical dementia patients compared to controls (Taylor et al., 2014; Roy et al., 2020). The expression of IRF7, a transcription factor regulating type-I IFN production, and itself an ISG, is strongly correlated with Braak stage and clinical progression. This elevated IRF7 expression is reflected at the protein level in the prefrontal cortex of AD patients (Taylor et al., 2014; Roy et al., 2020). Larger scale transcriptomic approaches in AD and other tauopathies also show that there is a complex signature of type-I IFN-mediated immune suppression and activation (Rexach et al., 2020). Genome wide association studies (GWAS) implicate genetic variation in innate immune response pathways as important contributors for AD and tau-associated dementias. Polymorphisms in several innate immune genes, including ISGs, are associated with AD risk (Salih et al., 2019). This includes OAS1, a cytosolic RNA sensor responsible for degrading cellular and viral RNAs (Magusali et al., 2021). A type-I IFN signature is therefore a key characteristic in AD and other tauopathies with a potential disease-modifying role.
Protein Pathologies and Aging Contribute to a Type-I IFN Signature
Protein assemblies are themselves agonists of the type-I IFN response. Aβ1−42 assemblies stimulate IFNα/β production in primary neurons, as well as in glial and choroid plexus epithelial cell cultures (Taylor et al., 2014; Mesquita et al., 2015; Minter et al., 2016). Pattern recognition receptors such as toll-like receptor 2 (TLR2) and TLR4, have been identified as receptors for assemblies of Aβ1−42 (Liu et al., 2012b; Hughes et al., 2020) and of phosphorylated tau (Meng et al., 2022). This signaling can elicit a downstream IFN response. Tau assemblies can further stimulate type-I IFN via a cytosolic microglial receptor, polyglutamine binding protein 1 (PQBP1) (Jin et al., 2021) and can activate STAT1 (Li et al., 2019) (Table 1). Protein assemblies may therefore be considered as endogenously-derived molecular patterns that provoke innate immune responses similar to PAMPs.

Table 1. Summary of studies addressing the production of type-I IFNs in ex vivo and in vitro models of AD and tauopathy.
In mouse models of Aβ pathology, transcriptomic analysis shows that there is an increased population of IFN-responsive microglia which highly express ISGs (Sala Frigerio et al., 2019; Dorman et al., 2021; Yang et al., 2021). Lineage tracing reveals that these ISG-expressing microglia accumulate progressively in amyloid disease models until a majority of microglia displays evidence of ISG expression (Roy et al., 2022). Similarly, for tau, animal models demonstrate a type-I IFN signature early in the neurodegenerative process (Rexach et al., 2020). Genetic deletion of IFNAR reduces the phagocytic capacity of microglia and dampens the production of pro-inflammatory cytokines in response to Aβ1−42. This suggests that type-I IFNs are produced and are important for mediating downstream clearance of aggregates and onward inflammatory events.
Aging itself has been shown to be associated with high levels of type-I IFN in the CNS. Baruch et al. report an age-dependent type-I IFN production at the choroid plexus (Baruch et al., 2014). This has a detrimental effect on cognition which can be reversed by anti-IFNAR antibody administration. Therefore, even in the absence of specific protein pathology, age-related effects contribute to a type-I IFN signature in the brain. These studies suggest that stimuli for the production of type-I IFN likely derive from multiple sources: age-related activation of innate immunity, protein aggregates engaging PRRs and, once disease is established, DAMPs arising from tissue damage related to neurodegeneration. The consequences of chronic type-I IFN production in the CNS on the development of further pathology are not fully elucidated with some apparently contradictory findings. However, accumulating evidence suggests that a IFN response, particularly when chronically produced, is a source of toxicity and potentially exacerbates protein aggregation, thereby setting in motion a destructive feed-forward loop.
The Role of Type-I IFNs in Disease Pathology
In the periphery, type-I IFNs provide potent protection against infection, yet lead to toxic consequences when over-produced. Dysregulated IFN production leads to severe disease states, exemplified by inherited interferonopathies which have symptoms that mirror viral infection (Crow and Stetson, 2021). In neurodegeneration, type-I IFN is emerging as a central mediator of cascading toxic consequences. Loss of type-I IFN signaling is protective in APP/PS1 mice and 5xFAD mice, alleviating synapse loss and microglial activation (Minter et al., 2016; Roy et al., 2020, 2022) (Table 2). Consistent with this model, administration of IFN-β to WT mice promotes microglial activation, neurotoxicity and synapse loss (Roy et al., 2020). In transgenic mice expressing human tau, cognitive impairment is ameliorated when STAT1 signaling is blocked (Li et al., 2019). Beyond a model of toxicity, further evidence suggests that type-I IFNs can promote further protein aggregation. In mouse models, β-amyloid pathology is ameliorated under conditions of Ifnar genetic deletion (Roy et al., 2020, 2022). The effect of IFN on tau aggregation remains unclear, though agonists of the IFN response such as LPS exacerbate pathology (Lee et al., 2010). This raises the prospect that an inappropriate innate immune response to protein aggregates sets in motion a destructive feed-forward loop by inducing further protein aggregation via type-I IFN.
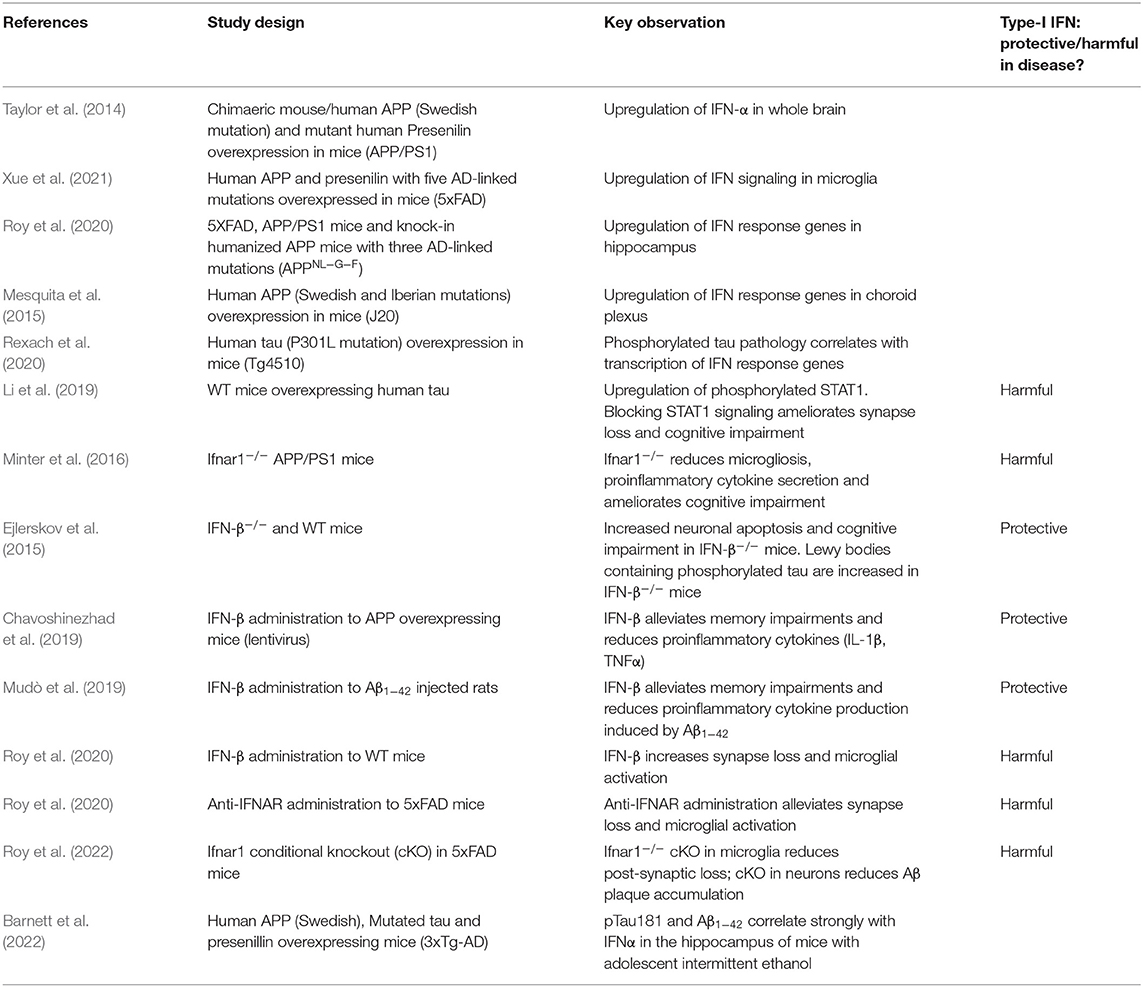
Table 2. Summary of studies evaluating type-I IFNs in in vivo models of AD and tauopathy.
Despite these multiple toxic effects of type-I IFN in the CNS, the view that its production is universally detrimental is not supported by other findings, particularly in acute settings. APP overexpression by lentiviral transduction or Aβ1−42 peptide delivery to the mouse brain induces pro-inflammatory cytokine production. This can be reduced by administration of IFN-β, consistent with the anti-inflammatory effects of type-I IFN (Chavoshinezhad et al., 2019; Mudò et al., 2019). Furthermore, genetic knockout of IFN-β is associated with reduced autophagic clearance and accumulation of ubiquitinated alpha-synuclein aggregates in mice (Ejlerskov et al., 2015). One possibility is that IFNs, while protective in acute settings, lead to damage when chronically over-produced, consistent with the “double-edged sword” hypothesis of innate immune activation in neurodegeneration (Wyss-Coray and Mucke, 2002). This would broadly align with our understanding of IFN in peripheral infection: that IFNs are highly protective when appropriately expressed yet can unleash severe damage when dysregulated or chronically over-produced.
Type-I IFNs in Models of Prion and Parkinson's Disease
Interesting parallels for tauopathies can be sought by examining other protein misfolding diseases of the CNS. Parkinson's disease (PD) is characterized by the aggregation of the cytosolic protein alpha-synuclein. Prion diseases such as Creutzfeld-Jakob disease in humans and scrapie in other animals are driven by the conversion of a membrane-anchored protein, PrP, to a misfolded variant. Genetic depletion of signaling components such as IRF3, Ifnar, TLR4 and TLR2 render mice more susceptible to the scrapie variant of PrP, PrPSc. This suggests a protective role for type-I IFNs in the recognition and control of prion assemblies (Ishibashi et al., 2012, 2019; Carroll et al., 2018). However, even here, type-I IFN production comes at a cost, as Nazmi et al. show that neuronal death is accelerated by Ifnar-dependent signaling (Nazmi et al., 2019).
In PD, type-I IFN and ISG transcripts are upregulated, similar to observations in AD and other tauopathies (Main et al., 2016). The effects of type-I IFN appear to be model-dependent. In one model of PD, neuronal loss is induced by 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) injection. Blockade of IFN signaling using anti-Ifnar antibodies supressed dopaminergic neuronal death, suggesting that type-I IFN signaling is neurotoxic (Main et al., 2016). In contrast, genetic deletion of IFN-β caused the formation of α-syn-positive Lewy body structures and reduced autophagic clearance (Ejlerskov et al., 2015). As in the tauopathies, these findings again point to production of type-I IFN having an important role in protection against protein aggregation, but with over-production contributing to toxicity. Any therapeutic intervention by manipulation of the type-I IFN pathway in proteopathies must therefore seek to target the over-production of IFN whilst ensuring that its essential functions in the control of proteinopathy are not unduly compromised.
Discussion
Type-I IFN plays a critical role in the brain during pathogen infection. Like classical pathogens, aggregated proteins including Aβ and tau can provoke an immune reaction that includes the production of type-I IFN. In vivo, the literature lacks clarity on whether IFNs are harmful or beneficial, likely owing to the timescales over which protection is measured. Protective effects of type-I IFN have been observed for Aβ, prion and α-syn especially in short-term challenge experiments, suggesting that common protective mechanisms may be at play. However, type-I IFNs promote downstream toxic consequences which may be amplified in a positive-feedback manner in response to ongoing tissue damage and further protein aggregation. Our understanding of these effects is in its infancy and remains largely without mechanistic detail. Further, for tau pathology, there remains little insight to the effect of IFN signaling due to the lack of studies using genetic knockout or experimental IFN-blockade. Future research should seek to dissect the IFN response at the level of specific ISGs to identify those that aid in limiting protein aggregation vs. those that promote toxic downstream consequences. An understanding at this level may allow selective pharmacological intervention to prevent the chronic toxic consequences of IFN signaling in neurodegeneration.
Author Contributions
SS and WM wrote and edited the manuscript. SS collated information used in the tables and supplementary tables. Both authors contributed to the article and approved the submitted version.
Funding
SS was funded by Alzheimer's Society Ph.D. Studentship, Grant Reference 488. WM was a Lister Institute Fellow and supported by a Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and the Royal Society (206248/Z/17/Z). This work was supported by the UK Dementia Research Institute, which receives its funding from DRI Ltd., funded by the UK Medical Research Council, Alzheimer's Society, and Alzheimer's Research UK.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2022.949340/full#supplementary-material
References
Barnett, A., David, E., Rohlman, A., Nikolova, V. D., Moy, S. S., Vetreno, R. P., et al. (2022). Adolescent binge alcohol enhances early alzheimer's disease pathology in adulthood through proinflammatory neuroimmune activation. Front. Pharmacol. 13, 884170. doi: 10.3389/fphar.2022.884170
Baruch, K., Deczkowska, A., David, E., Castellano, J. M., Miller, O., Kertser, A., et al. (2014). Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346, 89–93. doi: 10.1126/science.1252945
Braak, H., and Del Tredici, K. (2015). The preclinical phase of the pathological process underlying sporadic Alzheimer's disease. Brain 138, 2814–2833. doi: 10.1093/brain/awv236
Carroll, J. A., Race, B., Williams, K., and Chesebro, B. (2018). Toll-like receptor 2 confers partial neuroprotection during prion disease. PLoS ONE 13:e0208559. doi: 10.1371/journal.pone.0208559
Chavoshinezhad, S., Mohseni Kouchesfahani, H., Salehi, M. S., Pandamooz, S., Ahmadiani, A., and Dargahi, L. (2019). Intranasal interferon beta improves memory and modulates inflammatory responses in a mutant APP-overexpressing rat model of Alzheimer's disease. Brain Res. Bull. 150, 297–306. doi: 10.1016/j.brainresbull.2019.06.015
Clavaguera, F., Bolmont, T., Crowther, R. A., Abramowski, D., Frank, S., Probst, A., et al. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913. doi: 10.1038/ncb1901
Crow, Y. J., and Stetson, D. B. (2021). The type I interferonopathies: 10 years on. Nat. Rev. Immunol. doi: 10.1038/s41577-021-00633-9 [Epub ahead of print].
De Calignon, A., Polydoro, M., Suárez-Calvet, M., William, C., Adamowicz, D. H., Kopeikina, K. J., et al. (2012). Propagation of Tau pathology in a model of early Alzheimer's disease. Neuron 73, 685–697. doi: 10.1016/j.neuron.2011.11.033
Dorman, L., Nguyen, P., Escoubas, C., Vainchtein, I., Xiao, Y., Lidsky, P., et al. (2021). A type I interferon response defines a conserved microglial state required for effective neuronal phagocytosis. bioRxiv [Preprint]. doi: 10.1101/2021.04.29.441889
Ejlerskov, P., Hultberg, J. G., Wang, J., Carlsson, R., Ambjørn, M., Kuss, M., et al. (2015). Lack of neuronal IFN-β-IFNAR causes Lewy Body- and Parkinson's Disease-like Dementia. Cell 163, 324–339. doi: 10.1016/j.cell.2015.08.069
Frost, B., Jacks, R. L., and Diamond, M. I. (2009). Propagation of Tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 284, 12845–12852. doi: 10.1074/jbc.M808759200
Gerhard, A., Pavese, N., Hotton, G., Turkheimer, F., Es, M., Hammers, A., et al. (2006). In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiol. Dis. 21, 404–412. doi: 10.1016/j.nbd.2005.08.002
Goedert, M (2018). Tau filaments in neurodegenerative diseases. FEBS Lett. 592, 2383–2391. doi: 10.1002/1873-3468.13108
Goedert, M., and Spillantini, M. G. (2006). A century of Alzheimer's disease. Science 314, 777–781. doi: 10.1126/science.1132814
Goldmann, T., Blank, T., and Prinz, M. (2016). Fine-tuning of type I IFN-signaling in microglia–implications for homeostasis, CNS autoimmunity and interferonopathies. Curr. Opin. Neurobiol. 36, 38–42. doi: 10.1016/j.conb.2015.09.003
Guo, J. L., and Lee, V. M. Y. (2011). Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 286, 15317–15331. doi: 10.1074/jbc.M110.209296
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Hughes, C., Choi, M. L., Yi, J.-H., Kim, S.-C., Drews, A., George-Hyslop, P. S., et al. (2020). Beta amyloid aggregates induce sensitised TLR4 signalling causing long-term potentiation deficit and rat neuronal cell death. Commun. Biol. 3:79. doi: 10.1038/s42003-020-0792-9
Ishibashi, D., Atarashi, R., Fuse, T., Nakagaki, T., Yamaguchi, N., Satoh, K., et al. (2012). Protective role of interferon regulatory factor 3-mediated signaling against prion infection. J. Virol. 86, 4947–4955. doi: 10.1128/JVI.06326-11
Ishibashi, D., Homma, T., Nakagaki, T., Fuse, T., Sano, K., Satoh, K., et al. (2019). Type I interferon protects neurons from prions in in vivo models. Brain 142, 1035–1050. doi: 10.1093/brain/awz016
Iwasaki, A., and Medzhitov, R. (2004). Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5, 987–995. doi: 10.1038/ni1112
Jin, M., Shiwaku, H., Tanaka, H., Obita, T., Ohuchi, S., Yoshioka, Y., et al. (2021). Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 12:6565. doi: 10.1038/s41467-021-26851-2
Karran, E., and De Strooper, B. (2022). The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat. Rev. Drug Discov. 21, 306–318. doi: 10.1038/s41573-022-00391-w
Knopman, D. S., Amieva, H., Petersen, R. C., Chételat, G., Holtzman, D. M., Hyman, B. T., et al. (2021). Alzheimer disease. Nat. Rev. Dis. Primers 7:33. doi: 10.1038/s41572-021-00269-y
Laurent, C., Buée, L., and Blum, D. (2018). Tau and neuroinflammation: what impact for Alzheimer's Disease and Tauopathies? Biomed. J. 41, 21–33. doi: 10.1016/j.bj.2018.01.003
Le Bon, A., and Tough, D. F. (2002). Links between innate and adaptive immunity via type I interferon. Curr. Opin. Immunol. 14, 432–436. doi: 10.1016/S0952-7915(02)00354-0
Lee, D. C., Rizer, J., Selenica, M.-L. B., Reid, P., Kraft, C., Johnson, A., et al. (2010). LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J. Neuroinflammation 7:56. doi: 10.1186/1742-2094-7-56
Li, X.-G., Hong, X., Wang, Y.-L., Zhang, S.-J., Zhang, J., Li, X.-C., et al. (2019). Tau accumulation triggers STAT1-dependent memory deficits by suppressing NMDA receptor expression. EMBO Rep. 20:e47202. doi: 10.15252/embr.201847202
Liu, L., Drouet, V., Wu, J. W., Witter, M. P., Small, S. A., Clelland, C., et al. (2012a). Trans-synaptic spread of Tau pathology in vivo. PLoS ONE 7:e31302. doi: 10.1371/journal.pone.0031302
Liu, S., Liu, Y., Hao, W., Wolf, L., Kiliaan, A. J., Penke, B., et al. (2012b). TLR2 is a primary receptor for Alzheimer's Amyloid β Peptide to trigger neuroinflammatory activation. J. Immunol. 188, 1098–1107. doi: 10.4049/jimmunol.1101121
Magusali, N., Graham, A. C., Piers, T. M., Panichnantakul, P., Yaman, U., Shoai, M., et al. (2021). A genetic link between risk for Alzheimer's disease and severe COVID-19 outcomes via the OAS1 gene. Brain 144, 3727–3741. doi: 10.1093/brain/awab337
Main, B. S., Zhang, M., Brody, K. M., Ayton, S., Frugier, T., Steer, D., et al. (2016). Type-1 interferons contribute to the neuroinflammatory response and disease progression of the MPTP mouse model of Parkinson's disease. Glia 64, 1590–1604. doi: 10.1002/glia.23028
Malpetti, M., Passamonti, L., Rittman, T., Jones, P. S., Vázquez Rodríguez, P., Bevan-Jones, W. R., et al. (2020). Neuroinflammation and Tau Colocalize in vivo in Progressive Supranuclear Palsy. Ann. Neurol. 88, 1194–1204. doi: 10.1002/ana.25911
McEwan, W. A., Falcon, B., Vaysburd, M., Clift, D., Oblak, A. L., Ghetti, B., et al. (2017). Cytosolic Fc receptor TRIM21 inhibits seeded tau aggregation. Proc. Natl. Acad. Sci. U.S.A. 114, 574–579. doi: 10.1073/pnas.1607215114
Meng, J. X., Zhang, Y., Saman, D., Haider, A. M., De, S., Sang, J. C., et al. (2022). Hyperphosphorylated Tau self-assembles into amorphous aggregates eliciting TLR4-dependent responses. Nat. Commun. 13:2692. doi: 10.1038/s41467-022-30461-x
Mesquita, S. D., Ferreira, A. C., Gao, F., Coppola, G., Geschwind, D. H., Sousa, J. C., et al. (2015). The choroid plexus transcriptome reveals changes in type I and II interferon responses in a mouse model of Alzheimer's disease. Brain. Behav. Immun. 49, 280–292. doi: 10.1016/j.bbi.2015.06.008
Miller, L. V. C., Mukadam, A. S., Durrant, C. S., Vaysburd, M. J., Katsinelos, T., Tuck, B. J., et al. (2021). Tau assemblies do not behave like independently acting prion-like particles in mouse neural tissue. Acta Neuropathol. Commun. 9:41. doi: 10.1186/s40478-021-01141-6
Minter, M. R., Moore, Z., Zhang, M., Brody, K. M., Jones, N. C., Shultz, S. R., et al. (2016). Deletion of the type-1 interferon receptor in APPSWE/PS1DeltaE9 mice preserves cognitive function and alters glial phenotype. Acta Neuropathol. Commun. 4:72. doi: 10.1186/s40478-016-0341-4
Mudò, G., Frinchi, M., Nuzzo, D., Scaduto, P., Plescia, F., Massenti, M. F., et al. (2019). Anti-inflammatory and cognitive effects of interferon-β1a (IFNβ1a) in a rat model of Alzheimer's disease. J. Neuroinflammation 16:44. doi: 10.1186/s12974-019-1417-4
Navarro, V., Sanchez-Mejias, E., Jimenez, S., Muñoz-Castro, C., Sanchez-Varo, R., Davila, J. C., et al. (2018). Microglia in Alzheimer's disease: activated, dysfunctional or degenerative. Front. Aging Neurosci. 10:140. doi: 10.3389/fnagi.2018.00140
Nazmi, A., Field, R. H., Griffin, E. W., Haugh, O., Hennessy, E., Cox, D., et al. (2019). Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia 67, 1254–1276. doi: 10.1002/glia.23592
Nelson, P. T., Jicha, G. A., Schmitt, F. A., Liu, H., Davis, D. G., Mendiondo, M. S., et al. (2007). Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort. J. Neuropathol. Exp. Neurol. 66, 1136–1146. doi: 10.1097/nen.0b013e31815c5efb
Paulus, W., Bancher, C., and Jellinger, K. (1993). Microglial reaction in Pick's disease. Neurosci. Lett. 161, 89–92. doi: 10.1016/0304-3940(93)90147-D
Prinz, M., Schmidt, H., Mildner, A., Knobeloch, K.-P. P., Hanisch, U.-K. K., Raasch, J., et al. (2008). Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity 28, 675–686. doi: 10.1016/j.immuni.2008.03.011
Rexach, J. E., Polioudakis, D., Yin, A., Swarup, V., Chang, T. S., Nguyen, T., et al. (2020). Tau pathology drives dementia risk-associated gene networks toward chronic inflammatory states and immunosuppression. Cell Rep. 33:108398. doi: 10.1016/j.celrep.2020.108398
Roh, J. S., and Sohn, D. H. (2018). Damage-associated molecular patterns in inflammatory diseases. Immune Netw. 18:e27. doi: 10.4110/in.2018.18.e27
Roy, E. R., Chiu, G., Li, S., Propson, N. E., Kanchi, R., Wang, B., et al. (2022). Concerted type I interferon signaling in microglia and neural cells promotes memory impairment associated with amyloid β plaques. Immunity 55, 879–894.e6. doi: 10.1016/j.immuni.2022.03.018
Roy, E. R., Wang, B., Wan, Y., Chiu, G., Cole, A., Yin, Z., et al. (2020). Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Invest. 130, 1912–1930. doi: 10.1172/JCI133737
Sala Frigerio, C., Wolfs, L., Fattorelli, N., Thrupp, N., Voytyuk, I., Schmidt, I., et al. (2019). The major risk factors for Alzheimer's disease: age, sex, and genes modulate the microglia response to Aβ Plaques. Cell Rep. 27, 1293–1306.e6. doi: 10.1016/j.celrep.2019.03.099
Salih, D. A., Bayram, S., Guelfi, S., Reynolds, R. H., Shoai, M., Ryten, M., et al. (2019). Genetic variability in response to amyloid beta deposition influences Alzheimer's disease risk. Brain Commun. 1:fcz022. doi: 10.1093/braincomms/fcz022
Scheu, S., Ali, S., Mann-Nuttel, R., Richter, L., Arolt, V., Dannlowski, U., et al. (2019). Interferon beta-mediated protective functions of microglia in central nervous system autoimmunity. Int. J. Mol. Sci. 20:190. doi: 10.3390/ijms20010190
Serrano-Pozo, A., Mielke, M. L., Gómez-Isla, T., Betensky, R. A., Growdon, J. H., Frosch, M. P., et al. (2011). Reactive Glia not only associates with plaques but also parallels tangles in Alzheimer's disease. Am. J. Pathol. 179, 1373–1384. doi: 10.1016/j.ajpath.2011.05.047
Shaw, A. E., Hughes, J., Gu, Q., Behdenna, A., Singer, J. B., Dennis, T., et al. (2017). Fundamental properties of the mammalian innate immune system revealed by multispecies comparison of type I interferon responses. PLoS Biol. 15:e2004086. doi: 10.1371/journal.pbio.2004086
Spillantini, M. G., and Goedert, M. (2013). Tau pathology and neurodegeneration. Lancet Neurol. 12, 609–622. doi: 10.1016/S1474-4422(13)70090-5
Stelzmann, R. A., Norman Schnitzlein, H., and Reed Murtagh, F. (1995). An english translation of Alzheimer's 1907 paper, ‘Uber eine eigenartige erkankung der hirnrinde'. Clin. Anat. 8, 429–431. doi: 10.1002/ca.980080612
Taipa, R., das Neves, S. P., Sousa, A. L., Fernandes, J., Pinto, C., Correia, A. P., et al. (2019). Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer's disease and their correlation with cognitive decline. Neurobiol. Aging 76, 125–132. doi: 10.1016/j.neurobiolaging.2018.12.019
Tarkowski, E (2003). Intrathecal inflammation precedes development of Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry 74, 1200–1205. doi: 10.1136/jnnp.74.9.1200
Taylor, J. M., Minter, M. R., Newman, A. G., Zhang, M., Adlard, P. A., and Crack, P. J. (2014). Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer's disease. Neurobiol. Aging 35, 1012–1023. doi: 10.1016/j.neurobiolaging.2013.10.089
Wyss-Coray, T., and Mucke, L. (2002). Inflammation in neurodegenerative disease—A double-edged sword. Neuron 35, 419–432. doi: 10.1016/S0896-6273(02)00794-8
Xue, F., Tian, J., Yu, C., Du, H., and Guo, L. (2021). Type I interferon response-related microglial Mef2c deregulation at the onset of Alzheimer's pathology in 5xFAD mice. Neurobiol. Dis. 152, 105272. doi: 10.1016/j.nbd.2021.105272
Keywords: type-I interferon response, Alzheimer's disease, tauopathies, innate immunity, antiviral immunity, IFN, amyloid-beta pathology, tau pathology
Citation: Sanford SAI and McEwan WA (2022) Type-I Interferons in Alzheimer's Disease and Other Tauopathies. Front. Cell. Neurosci. 16:949340. doi: 10.3389/fncel.2022.949340
Received: 20 May 2022; Accepted: 16 June 2022;
Published: 15 July 2022.
Edited by:
Marta Pérez González, University College London, United KingdomReviewed by:
Paige Mumford, University College London, United KingdomCopyright © 2022 Sanford and McEwan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophie A. I. Sanford, ss2575@cam.ac.uk; William A. McEwan, wm305@cam.ac.uk